
First-in-Human Studies
The purpose of First-in-Human (FIH) trials is to determine a safe dose range for further clinical development, and to develop a better understanding of the investigational product’s pharmacology such that translational risk to humans can be minimized. Furthermore, these trials offer an opportunity to obtain crucial pharmacologic data which can inform prescribing physicians with information about the proper use and risks of the product once it has been approved for marketplace availability. Successful execution of FIH studies requires a cross-functional approach which takes into consideration preclinical safety testing, drug formulation, regulatory requirements, study design, and safety monitoring.
For over two decades, the team at Dr. Vince Clinical Research has performed First-in-Human clinical studies for both small and large molecules and vaccines across various therapeutic areas and routes of administration, affording us the expertise to aid biopharmaceutical sponsors in the successful planning and conduct of these studies during an investigational treatment’s development path.
DVCR’s First-in-Human Study Experience

Investigator team has participated in over 50 FIH trials
Leadership team averaging a decade of experience in early clinical development
Press Release covering recent First-in-Human study
Phase I Unit for First-in-Human Studies
Multiple aspects of our Phase 1 unit were designed with the goal of conducting successful First-in-Human studies.
- 90 beds for overnight confinement
- eSource/EDC with real-time data access for expedited dose escalation decisions
- Wall-mounted vital signs machines at each bedside to reduce data variability across subjects throughout the duration of the trial
- KIntegrated with eSource/EDC for automatic data upload
- KSame blood pressure cuff used throughout the duration of clinic stays for each subject
- KCalibrated annually by the manufacturer
- Strategically located crash carts and panic buttons
- Video surveillance in subject areas
- Utilization of Verified Clinical Trials to prevent dual subject enrollment
- Multiple nearby hospitals
- On-site cGMP pharmacy capable of handling and manipulating study drugs for various routes of administration
When considering investigational product preparation for FIH trials, early dialog between biopharmaceutical sponsors and CROs/Phase 1 sites is crucial in preventing potential study delays. For example, FIH clinical trials evaluating a biologic as the investigational product will need to undergo compatibility studies prior to submitting the Investigational New Drug (IND) application. In this instance, it is critical to utilize the same type of IV bag (e.g., DEHP-free), infusion set (with or without an in-line filter, DEHP-free, etc.) and infusion pump for the compatibility study as is planned for use in the First-in-Human trial. With over 50 years of combined experience, DVCR’s pharmacy team has the capability and bandwidth to procure these necessary and essential ancillary supplies and equipment through approved vendors.

Reach Out Today
If you would like to receive more information regarding how our CRO can be leveraged for your upcoming First in Human trial, please contact us.
Phase I Unit for First-in-Human Studies
Multiple aspects of our Phase 1 unit were designed with the goal of conducting successful First-in-Human studies.
- 90 beds for overnight confinement
- eSource/EDC with real-time data access for expedited dose escalation decisions
- Wall-mounted vital signs machines at each bedside to reduce data variability across subjects throughout the duration of the trial
- KIntegrated with eSource/EDC for automatic data upload
- KSame blood pressure cuff used throughout the duration of clinic stays for each subject
- KCalibrated annually by the manufacturer
- Strategically located crash carts and panic buttons
- Video surveillance in subject areas
- Utilization of Verified Clinical Trials to prevent dual subject enrollment
- Multiple nearby hospitals
- On-site cGMP pharmacy capable of handling and manipulating study drugs for various routes of administration
When considering investigational product preparation for FIH trials, early dialog between biopharmaceutical sponsors and CROs/Phase 1 sites is crucial in preventing potential study delays. For example, FIH clinical trials evaluating a biologic as the investigational product will need to undergo compatibility studies prior to submitting the Investigational New Drug (IND) application. In this instance, it is critical to utilize the same type of IV bag (e.g., DEHP-free), infusion set (with or without an in-line filter, DEHP-free, etc.) and infusion pump for the compatibility study as is planned for use in the First-in-Human trial. With over 50 years of combined experience, DVCR’s pharmacy team has the capability and bandwidth to procure these necessary and essential ancillary supplies and equipment through approved vendors.
Reach Out Today
If you would like to receive more information regarding how our CRO can be leveraged for your upcoming First in Human trial, please contact us.
FIH Study Design
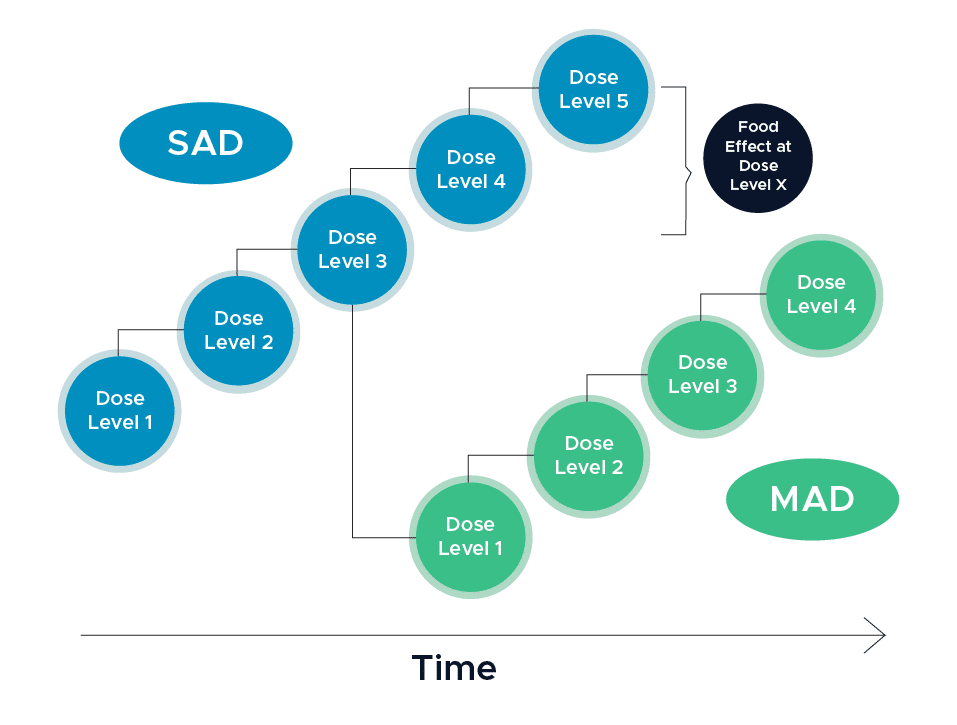
First-in-Human clinical trials are blinded, placebo-controlled, and typically enroll eight subjects per cohort randomized in a 3:1 ratio such that six subjects receive the active therapy and two subjects receive a placebo (1). In a single ascending dose (SAD) design, subjects will receive a single dose of the investigational treatment, and the dose level will be increased in each subsequent cohort assuming the study does not meet its predefined stopping criteria. The number of planned doses in a single ascending dose design is usually between three to eight cohorts.
Sponsors may elect to conduct a single ascending dose (SAD) and multiple ascending dose (MAD) study together in one protocol. In the multiple ascending dose cohorts, subjects will receive multiple doses of the investigational treatment and the dose level will be increased in each subsequent cohort so long as stopping criteria are not met. Multiple ascending dose cohorts are typically done in a group of 10 subjects. It is in these cohorts where patient populations might be enrolled instead of normal healthy volunteers in order to obtain early efficacy data on the product.
In addition to safety and dose ranging measures, biopharmaceutical companies may elect to incorporate assessments on food effect, relative bioavailability, QT prolongation, and drug interactions as a potential part of FIH trials. This being the case, the use of an adaptive trial design may be necessary in order to modify the study design when certain data is made available. For example, food effect assessments can be incorporated into the SAD arm of the study once dose escalation has reached an anticipated therapeutically relevant level (1).
In order to determine if the dose level can be increased, a formal dose escalation meeting takes place between each of the dose cohorts and planned for within the study timelines. At the start of the study, a dose escalation committee (DEC) will be established and typically involves, at minimum, the Principal Investigator (PI) and a Medical Monitor. The specifics of the dose escalation process will be outlined in the study’s dose escalation charter. Typically, the following data will be reviewed:
- Safety data through a certain post-dose timepoint for all available subjects within the dose cohort
- All adverse events reported to date
- PK data through a certain post-dose timepoint
- KWhen review of PK data is involved in dose escalation, additional consideration must be taken to account for the time necessary to ship, receive and process and analyze the samples, as well as report on the PK data. This will have an impact on the overall project timelines and the speed at which doses can be escalated.
At the conclusion of each dose escalation meeting, a form will be signed by designated representatives that summarizes the decision to move to the next dose cohort or not to dose escalate. This summary will be sent to the IRB. IRB approval is not needed to dose escalate unless the dose is being changed from what is already listed in the protocol and ICF. Typically, protocols will include standard language which allows for the flexibility to move to a lower dose, repeat the dose or stop the escalation.
FIH Study Design
First-in-Human clinical trials are blinded, placebo-controlled, and typically enroll eight subjects per cohort randomized in a 3:1 ratio such that six subjects receive the active therapy and two subjects receive a placebo. (1) In a single ascending dose (SAD) design, subjects will receive a single dose of the investigational treatment, and the dose level will be increased in each subsequent cohort assuming the study does not meet its predefined stopping criteria. The number of planned doses in a single ascending dose design is usually between three to eight cohorts.
Sponsors may elect to conduct a single ascending dose (SAD) and multiple ascending dose (MAD) study together in one protocol. In the multiple ascending dose cohorts, subjects will receive multiple doses of the investigational treatment and the dose level will be increased in each subsequent cohort so long as stopping criteria are not met. Multiple ascending dose cohorts are typically done in a group of 10 subjects. It is in these cohorts where patient populations might be enrolled instead of normal healthy volunteers in order to obtain early efficacy data on the product.
In addition to safety and dose ranging measures, biopharmaceutical companies may elect to incorporate assessments on food effect, relative bioavailability, QT prolongation, and drug interactions as a potential part of FIH trials. This being the case, the use of an adaptive trial design may be necessary in order to modify the study design when certain data is made available. For example, food effect assessments can be incorporated into the SAD arm of the study once dose escalation has reached an anticipated therapeutically relevant level. (1)
In order to determine if the dose level can be increased, a formal dose escalation meeting takes place between each of the dose cohorts and planned for within the study timelines. At the start of the study, a dose escalation committee (DEC) will be established and typically involves, at minimum, the Principal Investigator (PI) and a Medical Monitor. The specifics of the dose escalation process will be outlined in the study’s dose escalation charter. Typically, the following data will be reviewed:
- Safety data through a certain post-dose timepoint for all available subjects within the dose cohort
- All adverse events reported to date
- PK data through a certain post-dose timepoint
- KWhen review of PK data is involved in dose escalation, additional consideration must be taken to account for the time necessary to ship, receive and process and analyze the samples, as well as report on the PK data. This will have an impact on the overall project timelines and the speed at which doses can be escalated.
At the conclusion of each dose escalation meeting, a form will be signed by designated representatives that summarizes the decision to move to the next dose cohort or not to dose escalate. This summary will be sent to the IRB. IRB approval is not needed to dose escalate unless the dose is being changed from what is already listed in the protocol and ICF. Typically, protocols will include standard language which allows for the flexibility to move to a lower dose, repeat the dose or stop the escalation.
Sentinel Dosing
Many first-in-class and large molecule studies will incorporate a sentinel dose cohort for the initial dose level within the SAD and MAD study designs. Sentinel dosing may also be implemented for subsequent dose levels within a SAD and/or MAD study if warranted.
Sentinel dosing involves at least two study volunteers (one randomized to active study drug and one randomized to placebo). The remainder of the dose cohort will receive study drug or placebo a minimum of 24 hours after the sentinel cohort is dosed. The decision to dose the remainder of the cohort will be based on sentinel dosing being well tolerated. Progression to dosing the remainder of the cohort is typically made by the Principal Investigator based on the sentinel cohort’s safety data (e.g., Vital signs, ECGs, Physical Exams, Adverse Events) and is not part of a formal dose escalation committee. The Medical Monitor may be involved with the site’s PI in the decision to dose the remainder of the cohort upon review of sentinel safety and tolerability data.
An adequate number of study volunteers are screened and confirmed for admission to ensure that the sentinel cohort and remainder of the dose cohort are dosed on time. Study-specific informed consent language includes a description of the sentinel dose cohort design so study volunteers are informed that they may be part of the sentinel cohort.
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Sentinel Dosing
Many first-in-class and large molecule studies will incorporate a sentinel dose cohort for the initial dose level within the SAD and MAD study designs. Sentinel dosing may also be implemented for subsequent dose levels within a SAD and/or MAD study if warranted.
Sentinel dosing involves at least two study volunteers (one randomized to active study drug and one randomized to placebo). The remainder of the dose cohort will receive study drug or placebo a minimum of 24 hours after the sentinel cohort is dosed. The decision to dose the remainder of the cohort will be based on sentinel dosing being well tolerated. Progression to dosing the remainder of the cohort is typically made by the Principal Investigator based on the sentinel cohort’s safety data (e.g., Vital signs, ECGs, Physical Exams, Adverse Events) and is not part of a formal dose escalation committee. The Medical Monitor may be involved with the site’s PI in the decision to dose the remainder of the cohort upon review of sentinel safety and tolerability data.
An adequate number of study volunteers are screened and confirmed for admission to ensure that the sentinel cohort and remainder of the dose cohort are dosed on time. Study-specific informed consent language includes a description of the sentinel dose cohort design so study volunteers are informed that they may be part of the sentinel cohort.

Volunteer Recruitment in FIH Clinical Trials
As FIH studies will be the first instance of administering an investigational product to human subjects, strategic volunteer recruitment is of the utmost importance. The Inclusion and Exclusion criteria must be structured to ensure that only the best-suited subjects are included in the study. Thus, careful consideration should be given to this section of the protocol. Consulting with the medical personnel at the Phase 1 site for feedback before finalizing the protocol is strongly encouraged.
Furthermore, ensuring that subjects are not simultaneously enrolled in other ongoing clinical studies is critical for study volunteer safety and to prevent unanticipated drug interactions between other investigational products. In order to do this, DVCR employs the use of Verified Clinical Trial’s registry and biometric screening prior to consenting subjects. By taking this step, our site staff can help prevent dual trial enrollment.
Volunteer Recruitment in FIH Clinical Trials
As FIH studies will be the first instance of administering an investigational product to human subjects, strategic volunteer recruitment is of the utmost importance. The Inclusion and Exclusion criteria must be structured to ensure that only the best-suited subjects are included in the study. Thus, careful consideration should be given to this section of the protocol. Consulting with the medical personnel at the Phase 1 site for feedback before finalizing the protocol is strongly encouraged.
Furthermore, ensuring that subjects are not simultaneously enrolled in other ongoing clinical studies is critical for study volunteer safety and to prevent unanticipated drug interactions between other investigational products. In order to do this, DVCR employs the use of Verified Clinical Trial’s registry and biometric screening prior to consenting subjects. By taking this step, our site staff can help prevent dual trial enrollment.

Integrating Cardiac Assessments into FIH Studies
In 2015, the ICH E14 guidance was revised to allow concentration-corrected QT interval analysis to be performed on data generated from First-in-Human (FIH) studies to assess the effect of the NCE on the QT interval. This spurred the development of new methods to collect and assess these effects and gather sufficient data to satisfy regulatory requirements.
In this design, early precision QT assessments are incorporated into the design of a FIH, single-ascending dose and multiple ascending dose (SAD/MAD) trial. This approach differs from more common cardiac safety assessments performed in Phase 1, which is typically based on the principal investigator’s read of 12-lead ECG printouts. The early precision QT methodology leverages both concentration effect modelling, as well as high precision analysis of 10x more data than conventional measurement methods (2).
It is advised that at minimum, six to nine subjects at sufficiently high dose levels (plasma levels) are evaluated using the EPQT approach. Adding doses to achieve plasma concentrations above targeted therapeutic levels is helpful. Another item to consider is the number of subjects receiving placebo, typically pooled across several dose groups. A minimum number of 8 subjects on placebo is ideal. According to Clario, “it seems prudent to use at least 4 dose groups with 6/2 on active/placebo and with two of the doses higher than what is believed to be the therapeutic dose (2).”
Dr. Vince Clinical Research is committed to advancing medicine by leveraging technology as it relates to cardiac rhythm analysis and the potential of medications to affect the cardiac conduction cycle. DVCR has the expertise and equipment necessary to conduct these critical trials. Our contemporary approach to cardiac studies enables us to ensure that our pharmaceutical sponsors receive deliverables smoothly and accurately while keeping volunteer safety at the forefront of our efforts. Click here to learn more about our early QT expertise.
References
1 Shen, J., Swift, B., Mamelok, R., Pine, S., Sinclair, J., & Attar, M. (2018). Design and conduct considerations for first-in-human trials. Clinical and Translational Science, 12(1), 6–19. https://doi.org/10.1111/cts.12582
2 Darpo, B. (2022, April). The Early Precision QT Approach: Driving earlier assessments of cardiac safety and supporting regulatory change. Endpoint Technology Services for Clinical Trial Management | Clario. Retrieved April 2023, from https://clario.com/wp-content/uploads/2022/04/Clario_White-paper_EPQT-24MAR2022.pdf
